熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
當前證據表明,硫化氫(H?S)在腦功能中扮演重要角色,可能作為神經調質和細胞內信使發揮作用。在哺乳動物中樞神經系統(CNS)中,H?S由氨基酸半胱氨酸通過胱硫醚β-合成酶(CBS)催化生成,同時產生副產物絲氨酸(Ser)。由于CBS是一種鈣離子和鈣調蛋白依賴性酶,H?S的生物合成應受細胞內鈣離子濃度的急性調控。此外,S-腺苷甲硫氨酸(SAM)作為CBS的變構激活劑也參與調控。
過去十年在闡明H?S在生理和病理條件下的細胞水平作用方面取得重大進展。生理水平的H?S首次被證明選擇性刺激NMDA受體介導的電流。這種刺激促進海馬LTP誘導,但僅在弱強直刺激存在下。H?S單獨不誘導LTP,表明H?S主要在活躍突觸促進LTP(Abe and Kimura,1996;Kimura,2000)。H?S增強NMDA受體功能的潛在機制尚不清楚,盡管一種可能的途徑是通過氧化還原調節散布于神經元NMDA受體胞外域的巰基,這些巰基對氧化/還原劑敏感。許多內源性(如吡咯喹啉醌、硫辛酸、活性氧自由基、谷胱甘肽、二氫硫辛酸)和外源性分子(氰化物、氟吡汀)能夠氧化和還原NMDA受體(Dingledine et al.,1999),分別導致受體反應減弱和增強。因此,H?S可能憑借其還原特性激活NMDA受體。一個可能的氧化還原調節位點是位于NR1亞基胞外域的Cys對(Cys744和Cys798)(Sullivan et al.,1994)。
在細胞內,H?S通過cAMP生成增強NMDA受體介導的反應。外源性H?S增加大鼠原代大腦和小腦神經元培養物或某些神經元和膠質細胞系中cAMP生成(Kimura,2000)。cAMP在LTP啟動和晚期激活cAMP依賴性蛋白激酶(PKA)(Roberson and Sweatt,1996;Abel et al.,1997)。激活的PKA可能進而磷酸化NMDA受體亞基NR1、NR2A和NR2B的特定位點,以增強NMDA電流(這對LTP誘導至關重要)(Leonard and Hell,1997;Tingley et al.,1997)。因此,cAMP可能通過磷酸化NMDA受體調節LTP。除此之外,H?S還通過cAMP途徑以劑量依賴性方式減少對NMDA作出反應所需時間,即增加NMDA受體對其配體的敏感性。
最近還顯示,H?S上調γ-氨基丁酸(GABA)B受體(GABABR),這是一種位于突觸前和突觸后位點的G蛋白偶聯受體(Han et al.,2005)。刺激突觸后受體產生長時程抑制性突觸后電位,導致K?電導增加,對抑制性神經傳遞的微調很重要。H?S已顯示通過增加K?外流(可能通過ATP依賴性K?(KATP)通道)使CA1區和中縫背核神經元超極化(Reiffenstein et al.,1992)。在突觸前位點,GABABR通過抑制電壓敏感性Ca2?通道調節神經遞質(如GABA和Glu)釋放。H?S上調GABABR表達暗示H?S可能在維持腦內興奮/抑制平衡中起作用。
除其神經調節作用外,H?S已被證明在細胞外和細胞內微環境中保護神經元免受氧化應激。公認還原型谷胱甘肽(GSH)是腦中重要的抗氧化防御。它通過清除自由基和其他活性物質、去除過氧化氫和脂質過氧化物、防止生物分子氧化來保護大腦(Wu et al.,2004綜述)。H?S與GSH具有相似的神經保護特性,在體外具有相當的效力。這已通過H?S的能力得到證明:(i)抑制次氯酸介導的氧化損傷(Whiteman et al.,2005);(ii)抑制過氧亞硝酸鹽介導的蛋白質硝化和細胞毒性(Whiteman et al.,2004)。此外,H?S在體外輕易清除過氧化氫(H?O?)(Geng et al.,2004),后者是大多數細胞氧化應激的重要來源。盡管神經元(和膠質細胞)內谷胱甘肽水平在毫摩爾濃度范圍,但腦內其細胞外水平幾乎為零(Halliwell,2001;Bayir et al.,2002;Whiteman et al.,2005)。因此,細胞外環境高度依賴其他穩態調節的非谷胱甘肽抗氧化劑(如抗壞血酸)清除自由基(Rice,2000)。因此,由于其高內源性生成、易擴散特性和與GSH相當的強抗氧化效力,H?S可能成為腦細胞外微環境中另一種重要的內源性抗氧化劑候選物。
H?S增加神經元中還原型谷胱甘肽(GSH)的生成(Kimura and Kimura,2004)。NaHS處理本身能夠通過增強γ-谷氨酰半胱氨酸合成酶(γ-GCS)活性和上調Cys(GSH合成的限速底物)轉運來增加谷胱甘肽量。這種谷胱甘肽含量的增加被證明保護神經元免受氧毒性(一種由氧化應激引發的程序性細胞死亡),后者由高濃度Glu觸發。H?S還通過激活ATP依賴性K?(KATP)和Cl?通道以及增加谷胱甘肽水平,保護永生化小鼠海馬細胞系細胞免受氧化性Glu毒性。通過這兩種機制,H?S能夠在不同類型的神經細胞中提供對Glu誘導細胞死亡的完全保護,將細胞活力提高至未用Glu處理的神經元相似水平(Kimura and Kimura,2004;Kimura et al.,2006)。
膠質細胞
H?S在膠質細胞中扮演重要的神經調節作用。星形膠質細胞作為膠質細胞的主要類型,在維持神經元興奮性、調節腦pH穩態以及攝取突觸周圍的多種神經遞質(包括谷氨酸)中發揮關鍵作用(Koehler et al.,2006)。更重要的是,星形膠質細胞內鈣離子濃度([Ca2?]i)的充分增加能夠誘導并傳播至鄰近星形膠質細胞的擴散性鈣離子升高波,稱為“鈣波”(Dani et al.,1992)。與神經元通過產生動作電位傳遞信號不同,星形膠質細胞和其他膠質細胞通過鈣信號相互通訊(Braet et al.,2004綜述)。這為星形膠質細胞作為合胞體調節神經元和血管功能提供了基礎,暗示膠質細胞在突觸傳遞中的整體調節作用(Braet et al.,2004;Koehler et al.,2006)。外源性H?S可在原代星形膠質細胞培養物和海馬切片中引發鈣波(Nagai et al.,2004)。這種由H?S觸發的鈣波之前會伴隨[Ca2?]i的升高,該升高通過質膜上的鈣通道介導鈣內流,并在較小程度上通過細胞內鈣庫的鈣釋放實現。在腦切片中,H?S引起的[Ca2?]i增加會擴散至鄰近星形膠質細胞群并觸發鈣波。
與小膠質細胞不同,小膠質細胞作為常駐中樞神經系統的巨噬細胞群體,在外來挑戰時可被激活,類似于外周巨噬細胞(Farber and Kettenmann,2005綜述)。小膠質細胞在阿爾茨海默病(AD)(Wojtera et al.,2005)和帕金森病(Kim and Joh,2006)等神經元疾病的進展中被認為發揮作用。我們近期發現,外源性H?S應用可顯著提高小膠質細胞的[Ca2?]i,且呈劑量依賴性(Lee et al.,2006)。外源性H?S通過質膜介導鈣內流以及細胞內鈣庫釋放鈣離子觸發鈣內流。這種內流部分依賴于腺苷酸環化酶的激活,且獨立于磷脂酶C-蛋白激酶C-三磷酸肌醇通路。此外,抑制內源性H?S合成顯著降低[Ca2?]i,表明內源性H?S可能對[Ca2?]i穩態具有正向調節作用。除作為第二信使外,鈣離子作為整合因子控制小膠質細胞在靜息和激活狀態下的行為,其中基礎鈣濃度升高是脂多糖(LPS)刺激后激活小膠質細胞的特征(Hoffmann et al.,2003)。憑借H?S的易擴散特性,可以合理推測H?S可能通過升高鄰近小膠質細胞的基礎鈣水平在激活它們中發揮作用。
神經炎癥是一種由促炎細胞因子介導的過程,可由全身組織損傷引發,但最常見的是與神經系統直接損傷相關。神經炎癥涉及激活免疫細胞、膠質細胞和神經元的神經-免疫相互作用(Myers et al.,2006)。目前認識到所有主要的神經病理狀態均以顯著的炎癥反應為特征,這些反應主要由膠質細胞介導。激活的膠質細胞產生各種促炎或抗炎趨化因子,被認為對啟動和引導免疫細胞浸潤至腦組織并協調其活動至關重要(Morale et al.,2006)。小膠質細胞是駐留于中樞神經系統的特化巨噬細胞,其在神經炎癥中的激活作用至關重要(Moore and O'Banion,2002;Craft et al.,2005)。激活的小膠質細胞產生并釋放某些促炎因子(如一氧化氮、腫瘤壞死因子-α(TNF-α)和白介素-1β(IL-1β)),這些因子進一步加劇組織損傷并導致細胞死亡(Wojtera et al.,2005)。Hu等(2007)近期報道,H?S通過顯著減弱LPS刺激的膠質細胞中一氧化氮和TNF-α分泌,以濃度依賴性方式保護小膠質細胞和星形膠質細胞免受LPS誘導的炎癥反應。其機制似乎涉及抑制LPS誘導的誘導型一氧化氮合酶(iNOS)表達和p38絲裂原活化蛋白激酶(MAPK)磷酸化。
盡管有證據表明H?S對神經元具有神經保護作用,但迄今為止尚未開展關于H?S對膠質細胞群體保護作用的研究。多項研究證明谷胱甘肽(GSH)優先定位于膠質細胞(Slivka et al.,1987;Raps et al.,1989),膠質細胞中平均細胞內水平為4 mM,而神經元中為2.5 mM(Rice and Russo-Menna,1998)。鑒于H?S在神經元中增加GSH含量的作用,H?S可能對膠質細胞發揮相似甚至更強的效應以增強抗氧化性GSH。
生理功能
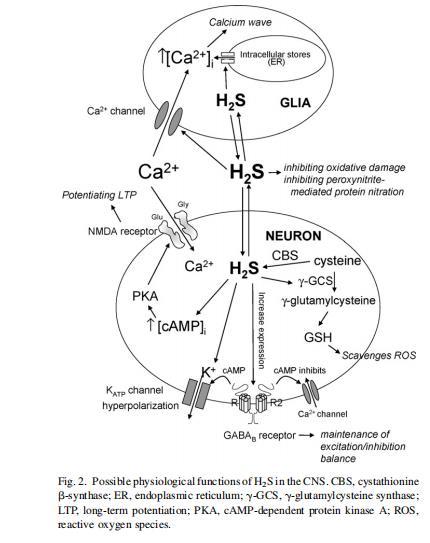
基于上述H?S的已知作用,其在腦中的潛在生理功能可能包括鈣穩態調節、LTP增強、氧化應激抑制以及神經傳遞調控(圖2)。
 相關新聞
相關新聞